Publications
Cancer-associated fibroblast subtypes differentially modulate natural killer cells in cancer
Rodrigues LN, Munir H, Thate Arrazola I, Gaida MM, Eckert C, Boulanger J, Ferreira ACF, McKenzie ANJ, Shields JD.
in: Cell Reports. 2026
Abstract
Natural killer (NK) cells are cytotoxic innate lymphoid cells which directly kill tumor cells, thus represent an attractive target for immunotherapy. However, NK cells face immunosuppression in the tumor microenvironment (TME), rendering them dysfunctional. While cancer-associated fibroblasts (CAFs) represent an abundant, heterogeneous component of pancreatic ductal adenocarcinoma (PDAC), their interplay with NK cells is largely understudied. Analyzing human samples and employing mouse models of PDAC and functional assays, we observed that intratumoral NK cells are immature, and TGF-β driven myofibroblastic (my)CAFs are strong NK suppressors, in contrast to inflammatory (i)CAF. Furthermore, myCAF-enriched tumor areas excluded NK cells, consistent with their reduced capacity to attract NK cells. Pancreatic CAFs in general reduced NK cell cytotoxicity by direct contact and via soluble factors, including prostaglandin E2 (PGE2). This work reveals distinct and overlapping roles of CAF subpopulations on NK cell functions, suggesting that overcoming CAF-imposed barriers to NK cytotoxicity and tumor infiltration is essential to unleash their anti-tumoral properties.
Keywords: CAF; CP: cancer; CP: immunology; NK cells; PDAC; PGE2; fibroblasts; tumor microenvironment.
Copyright © 2026 MRC Laboratory of Molecular Biology. Published by Elsevier Inc. All rights reserved.
Regulation of immune checkpoint molecules in cancer immune evasion and therapy
Eris C, Zu C, Xiao Y, Platten M, Sun C
in: Nat Rev Cancer. 2026
Abstract
Immune checkpoint molecules are essential regulators of immune homeostasis, maintaining the balance between activation and tolerance. In cancer, tumours exploit checkpoint pathways to suppress antitumour immunity and promote progression. The advent of immune checkpoint inhibitors, particularly those that target the clinically validated PDL1-PD1 and CTLA4 axes, has transformed cancer therapy, and the LAG3 axis has recently entered clinical practice, yet most patients experience limited or transient benefit, often because the checkpoint molecules become dysregulated. Here, we examine how multilayered regulatory mechanisms operating at the genetic, epigenetic, transcriptional, post-transcriptional, translational and post-translational levels collectively shape checkpoint abundance and function in tumour and immune cells. We further connect these regulatory processes to immune evasion and therapeutic resistance and highlight how this knowledge informs biomarker development and mechanism-guided strategies to improve immunotherapy outcomes.
© 2026. Springer Nature Limited.
Protocol for natural and biomedical image processing in the hypercomplex domain using the 2D orthogonal planes split
Valous NA, Hitzer E, Duşe D, Rojas Moraleda R, Popp F, Suarez-Carmona M, Berthel A, Papageorgiou I, Fremd C, Rölle A, Westhoff CC, Lenoir B, Halama N, Zörnig I, Jäger D.
in: Nat Rev Cancer. 2026
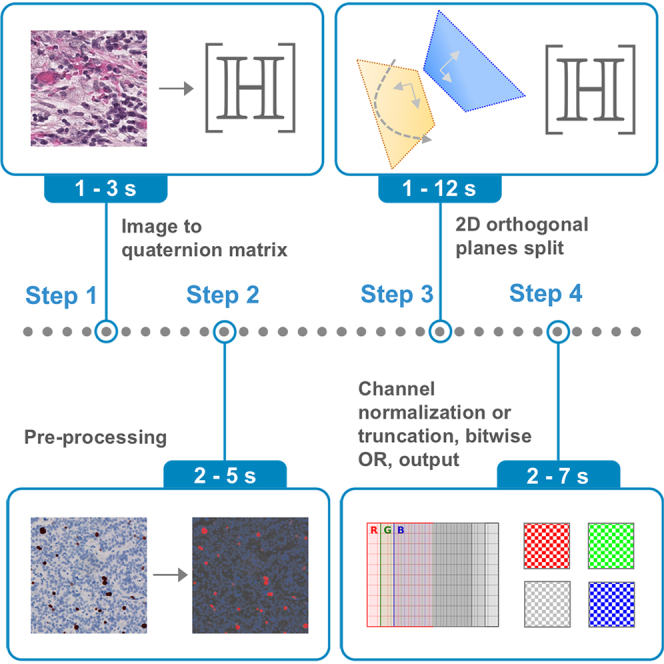
Abstract
Immune checkpoint molecules are essential regulators of immune homeostasis, maintaining the balance between activation and tolerance. In cancer, tumours exploit checkpoint pathways to suppress antitumour immunity and promote progression. The advent of immune checkpoint inhibitors, particularly those that target the clinically validated PDL1-PD1 and CTLA4 axes, has transformed cancer therapy, and the LAG3 axis has recently entered clinical practice, yet most patients experience limited or transient benefit, often because the checkpoint molecules become dysregulated. Here, we examine how multilayered regulatory mechanisms operating at the genetic, epigenetic, transcriptional, post-transcriptional, translational and post-translational levels collectively shape checkpoint abundance and function in tumour and immune cells. We further connect these regulatory processes to immune evasion and therapeutic resistance and highlight how this knowledge informs biomarker development and mechanism-guided strategies to improve immunotherapy outcomes.
© 2026. Springer Nature Limited.
Harnessing lipid-driven immunometabolic pathways in omental metastases to enhance immunotherapy in patients with ovarian cancer
Suarez-Carmona M, Hampel M, Zhang XW, Pöchmann A, Grauling-Halama SA, Valous NA, Charoentong P, Ferber D, Wissfeld J, Höflich A, Goriely S, Detavernier A, Azouz A, Rongvaux A, Zukunft S, Fleming I, Okun JG, Baracos V, Heikenwalder M, Zitvogel L, Xu X, Xu C, Volkmar M, Schraivogel D, Steinmetz L, Hamanishi J, Mandai M, Gaida M, Mokry T, Nattenmüller J, Sedlaczek O, Monje N, Schwab R, Hasenburg A, Mavratzas A, Boger RJ, Marmé F, Schott S, Halama N.
in: Sig Transduct Target Therapy. 2026
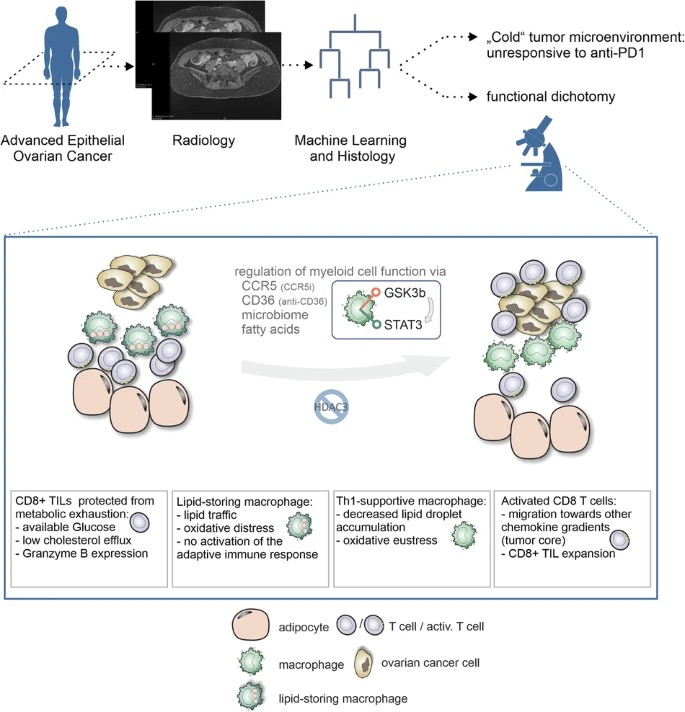
Abstract
Immunotherapy with immune checkpoint blockade (ICB) in epithelial ovarian carcinoma (EOC) shows limited clinical benefit only for a small subset of patients. Overall response rates are low, so that overcoming immunotherapy resistance and improved stratification are key. In this study, we investigated the immunometabolic landscape of EOC with a focus on omental metastases, identifying lipid-laden macrophages as central elements for actionable therapeutic vulnerabilities and giving rise to biomarkers for improved patient stratification. Using patient-derived explants, we demonstrated a functional dichotomy inside the typically lipid-rich microenvironment of omental metastases: augmented maintenance of effector T cell function, while lipid uptake and processing by tumor-associated macrophages (TAMs) induces oxidative stress–dependent signaling programs, which drive macrophage dysfunction and immune suppression. Pharmacological modulation of lipid-driven signaling pathways through CCR5 inhibition (inflammation modulation through maraviroc) or blockade of the lipid scavenger receptor CD36 reprograms TAMs, restores T cell activity, and enhances antitumor immune responses within lipid-rich tumor niches. Mechanistically, studies in humanized mouse models reveal that maraviroc-mediated CCR5 inhibition induces transcriptional programs associated with immune activation in stressed, lipid-laden human TAMs. Consistent with these mechanistic insights, we demonstrated that the specific immunometabolic niche in omental metastases is clinically associated with responsiveness to ICB. We propose a non-invasive radiomics and machine-learning–based analysis of imaging data to assess omental involvement for patient stratification.
Computational workflows for natural and biomedical image processing based on hypercomplex algebras
Valous NA, Hitzer E, Duşe D, Moraleda RR, Popp F, Suarez-Carmona M, Berthel A, Papageorgiou I, Fremd C, Rölle A, Westhoff CC, Lenoir B, Halama N, Zörnig I, Jäger D.
in: Patterns (N Y). 2025

The bigger picture
Images surround us, in photography, art, medicine, and science, but the way computers process them is often limited by how colors are mathematically represented. This work introduces a fresh approach based on quaternions, a special type of number system that treats each pixel as more than just three separate color values. Instead, colors are encoded in a geometric way, making it possible to handle images more naturally. By splitting each pixel into orthogonal planes, our framework provides new tools for transforming and analyzing images. We show how this can recolor or simplify pictures, enhance contrast in both everyday scenes and medical scans, and even digitally restain tissue samples in pathology, revealing structures that may otherwise remain hidden. Importantly, these workflows do not depend on training large AI models; instead, they use straightforward mathematical operations that are transparent and efficient. The impact is 2-fold. For everyday images, this method allows alternative visualizations, from new color renditions to effective grayscale conversions. For biomedical applications, it provides practical ways to enhance diagnostic images, separate overlapping stains in tissue sections, and improve pipelines for machine/deep learning. Because the computations scale efficiently with image size, the approach is also suitable for digital photography and whole-slide pathology scans. Overall, this study highlights how a mathematically rich but accessible framework can yield simple, fast, and versatile tools with real-world value. Bridging mathematics with practical image analysis opens a path toward more interpretable and reliable computational methods across both visual media and digital medicine.
Abstract: Summary
Quaternions, a type of hypercomplex number, can be applied to handling three-dimensional data, i.e., color images. Here, we demonstrate, by leveraging quaternions and the two-dimensional orthogonal planes split framework, image processing workflows for natural and biomedical images, including natural and biomedical image recolorization, natural image decolorization, natural and biomedical image contrast enhancement, and computational restaining and stain separation in histological images. We also demonstrate performance gains in machine learning and deep learning pipelines for histological images. The proposed workflows can regulate color appearance and image contrast, be part of automated processing pipelines, and assist in digital pathology applications. Employing basic arithmetic and matrix operations, this work offers a computationally accessible methodology that showcases versatility and consistency across processing tasks and a range of computer vision and biomedical applications. The proposed non-data-driven methods achieve comparable or better results to those reported in the literature, showcasing the potential of robust theoretical frameworks with practical effectiveness.
Keywords: quaternions; 2D orthogonal planes split; color images; computer vision; computational biomedicine
Benchmarking Software for DDA-PASEF Immunopeptidomics
Chen Y, Preikschat A, Arnold A, Pecori R, Gomez-Zepeda D, Tenzer S.
in: Molecular & Cellular Proteomics. 2025
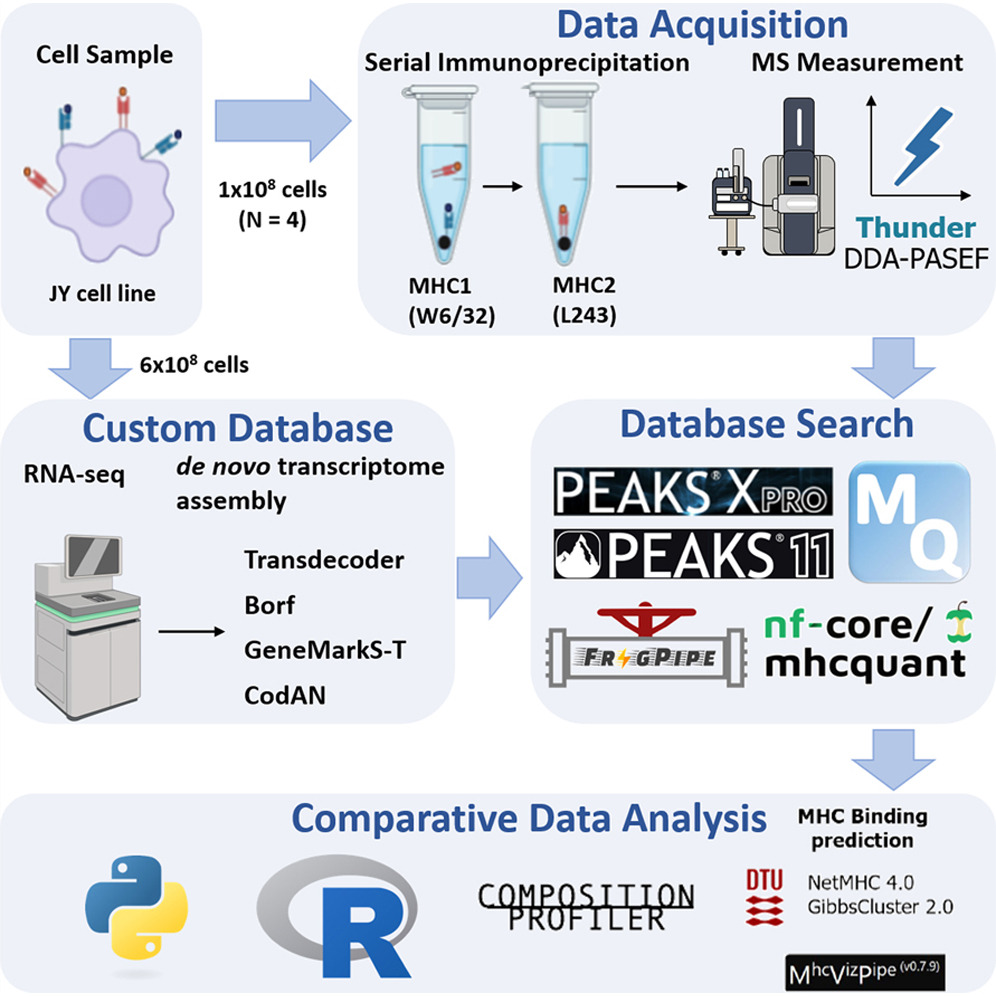
Abstract
Mass spectrometry (MS) is the method of choice for high-throughput identification of immunopeptides, which are generated by intracellular proteases, unlike proteomics peptides that are typically derived from trypsin-digested proteins. Therefore, the searching space for immunopeptides is not limited by proteolytic specificity, requiring more sophisticated software algorithms to handle the increased complexity. Despite the widespread use of MS in immunopeptidomics, there is a lack of systematic evaluation of data processing software, making it challenging to identify the optimal solution. In this study, we provide a comprehensive benchmarking of the most widespread/used data-dependent acquisition (DDA)-based software platforms for immunopeptidomics: MaxQuant, FragPipe, PEAKS and MHCquant. The evaluation was conducted using data obtained from the JY cell line using the Thunder-DDA-PASEF method. We assessed each software’s ability to identify immunopeptides and compared their identification confidence. Additionally, we examined potential biases in the results and tested the impact of database size on immunopeptide identification efficiency. Our findings demonstrate that all software platforms successfully identify the most prominent subset of immunopeptides with 1% false discovery rate (FDR) control, achieving medium to high identification confidence correlations. The largest number of immunopeptides were identified using the commercial PEAKS software, which is closely followed by FragPipe, making it a viable non-commercial alternative. However, we observed that larger database sizes negatively impacted the performance of some software platforms more than others. These results provide valuable insights into the strengths and limitations of current MS data processing tools for immunopeptidomics, supporting the immunopeptidomics/MS community in determining the right choice of software.
© 2025 The Authors. Published by Elsevier Inc on behalf of American Society for Biochemistry and Molecular Biologyé
Mainzelliste: Ten years of pseudonymization, record linkage, and informed consent management.
Tremper G, Brenner T, Ben Amor M, Kussel T, Lablans M.
in: Patterns. 2025
Abstract
Record linkage and pseudonymization are crucial tasks in collaborative biomedical research. Data for a patient are rarely stored in one place and therefore often need to be linked and integrated across multiple institutions. Mainzelliste is an open-source software solution designed to solve these challenges by providing a comprehensive and flexible toolkit for pseudonymization, record linkage, and consent management. It supports a variety of pseudonyms, record linkage methods, and modular, informed patient consents. A highly flexible REST application programming interface (API) allows tight integration into existing applications and workflows. Since its initial release in 2015, Mainzelliste has evolved into a vibrant open-source software solution “by researchers, for researchers” including a user-friendly graphical interface, support for HL7 FHIR for consent and patient data, and record linkage based on secure multi-party computation, thereby supporting secure and efficient biomedical research. © 2025 The Author(s).
Author keywords
informed consent management; pseudonymization; record linkage
© Copyright 2025 Elsevier B.V., All rights reserved.
Multicenter evaluation of label-free quantification in human plasma on a high dynamic range benchmark set
Distler U, Yoo H B, Kardell O, Hein D, Sielaff M, Scherer M, Jozefowicz A M, Leps C, Gomez-Zepeda D, von Toerne C, Merl-Pham J, Barth T K, Tüshaus J, Giesbertz P, Müller T, Kliewer G, Aljakouch K, Helm B, Unger H, Frey D L, Helm D, Schwarzmüller L, Popp O, Qin D, Wudy S I, Sinn L R, Mergner J, Ludwig C, Imhof A, Kuster B, Lichtenthaler S F, Krijgsveld J, Klingmüller U, Mertins P, Coscia F, Ralser M, Mülleder M, Hauck SM, Tenzer S
in: Nature Communications. 2025
Abstract
Human plasma is routinely collected during clinical care and constitutes a rich source of biomarkers for diagnostics and patient stratification. Liquid chromatography-mass spectrometry (LC-MS)-based proteomics is a key method for plasma biomarker discovery, but the high dynamic range of plasma proteins poses significant challenges for MS analysis and data processing. To benchmark the quantitative performance of neat plasma analysis, we introduce a multispecies sample set based on a human tryptic plasma digest containing varying low level spike-ins of yeast and E. coli tryptic proteome digests, termed PYE. By analysing the sample set on state-of-the-art LC-MS platforms across twelve different sites in data-dependent (DDA) and data-independent acquisition (DIA) modes, we provide a data resource comprising a total of 1116 individual LC-MS runs. Centralized data analysis shows that DIA methods outperform DDA-based approaches regarding identifications, data completeness, accuracy, and precision. DIA achieves excellent technical reproducibility, as demonstrated by coefficients of variation (CVs) between 3.3% and 9.8% at protein level. Comparative analysis of different setups clearly shows a high overlap in identified proteins and proves that accurate and precise quantitative measurements are feasible across multiple sites, even in a complex matrix such as plasma, using state-of-the-art instrumentation. The collected dataset, including the PYE sample set and strategy presented, serves as a valuable resource for optimizing the accuracy and reproducibility of LC-MS and bioinformatic workflows for clinical plasma proteome analysis.
© 2025. The Author(s).
Ganglioside Profiling Uncovers Distinct Patterns in High-Risk Neuroblastoma
Paret C, Wingerter A, Seidmann L, Ustjanzew A, Sathyamurthy S, Ludwig J, Schwickerath P, Brignole C, Pastorino F, Wagner S, El Malki K, Roth W, Sandhoff R, Faber J.
in: International Journal of Molecular Sciences. 2025
Abstract
High-risk (HR) neuroblastoma (NBL) patients often receive standardized treatment despite wide variations in clinical outcomes, underscoring the need for improved stratification tools. A distinguishing feature of NBL is the patient-specific expression of gangliosides (GGs), particularly GD2, which may serve as biomarkers. We analyzed GG profiles in 18 patient-derived tumors and 11 NBL cell lines using thin-layer chromatography and mass spectrometry. Expression of 0-, a-, and b-series GGs was examined and correlated with clinical risk, outcome, and gene expression data. Low-risk (LR) tumors expressed higher levels of complex b-series GGs. In HR tumors, five GG profiles (A-E) were identified. Profile A featured complex b-series GGs; B showed GD2 dominance; C showed synthesis arrest at GM3 or GD3 due to low expression of the GM2/GD2 synthase, encoded by the B4GALNT1 gene; D included complex a- and b-series GGs; and E was marked by GM2 and GD1a prevalence. B4GALNT1 expression served as a prognostic marker. Relapsed tumors following anti-GD2 therapy typically exhibited reduced GD2 levels, except for one profile A tumor that displayed a ceramide anchor shorter than those found in LR tumors. Astonishingly, the ceramide anchor composition of GD2 itself appears to separate LR and HR NBL, hinting at a role of ceramide synthases in NBL biology. All cell lines expressed GM2, but exhibited very low levels of complex b-series GGs. Profile C was found only in cell lines of the mesenchymal subtype. These findings support further investigation of GG composition and associated enzyme expression as potential biomarkers for risk stratification and treatment response in NBL.
Keywords: B4GALNT1; GD2; ceramide; dinutuximab; gangliosides; naxitamab; neuroblastoma.
Pancreatic resection with perioperative drug repurposing of propranolol and etodolac - the phase II randomized controlled PROSPER trial
Hüttner FJ, Klotz R, Giese NA, Kong B, Ahmed A, Merz D, Pöchmann A, Burghaus I, Hackert T, Strobel O, Mihaljevic AL, Michalski CW, Büchler MW, Diener MK
in: Langenbeck's Archives of Surgery. 2025
Abstract
Purpose: The perioperative period is characterized by psychological stress and inflammatory reactions that can contribute to disease recurrence or metastatic spread. These reactions are mediated particularly by catecholamines and prostaglandins. The PROSPER trial aimed to evaluate whether a perioperative drug repurposing with a non-selective betablocker (propranolol) and a COX-2 inhibitor (etodolac) is feasible and safe in the setting of pancreatic cancer surgery.
Methods: Patients undergoing partial pancreatoduodenectomy for pancreatic cancer were randomized to perioperative treatment with propranolol and etodolac or placebo. Main safety endpoint was the rate of serious adverse events (SAE) and the main feasibility endpoint was adherence. Overall and disease-free survival (DFS) as well as recurrences were assessed as efficacy parameters and the trial was accompanied by a translational study.
Results: The trial was prematurely closed due to slow recruitment. 26 patients were randomized, but 6 never started trial medication. Finally, 9 patients received the trial medication and 11 patients placebo. There were 6 SAE in the treatment vs. 14 in the placebo group. Adherence was lower in the treatment group, but without statistically significance. Median DFS was 16.36 months (95%-CI 1.18 - not reached) in verum vs. 11.25 (95%-CI 2.2 - 17.25) in placebo group. The rate of distant recurrences was 11.1% in verum vs. 54.5% in placebo group.
Conclusion: There were no safety concerns, but the trial intervention was not feasible given slow recruitment and limited adherence. However, the translational study and preliminary efficacy data revealed some promising findings, warranting further investigation.
De novo induction of tertiary lymphoid structures: an immunotherapeutic strategy in pancreatic cancer
Ahmed A, Springfeld C, Halama N
in: Signal Transduction and Targeted Therapy. 2025

Overview with central proposed elements of tertiary lymphoid structure (TLS) lymphoneogenesis in mouse models. IL-33 produced in the gut of PDAC-bearing mice leads to migration of KLRG1+ ILC2 cells towards the tumor and interaction with CD11b+ myeloid helper cells. This in turn attracts B cells (and T cells) to form TLSs (as shown in the schematic pixelated TLS representation). The induction of TLSs was recapitulated in mice treated with the human drug candidate H-rIL-33.
The role of the extracellular matrix protein SPOCK2 for bone physiology and hematopoiesis
Kumar R, Das S, Minka W, Reiter C, Pereira R, Fuhrmann D, Schneider R, Seshire A, Reusch C, Conche C, Imkeller K, Pajevic PD, Krause DS
in: Bone. 2025
Abstract
The bone marrow microenvironment (BMM) consists of different cellular and acellular components. These components synergize in regulating the process of hematopoiesis. Various extracellular matrix proteins are found amongst the acellular components. Secreted protein acidic and rich in cysteine (SPARC) is amongst the most abundant glycoproteins in bone. Sparc/osteonectin, cwcv, and Kazal-like domains proteoglycan 2 (SPOCK2) is a member of the SPARC family, and its role in bone metabolism and hematopoiesis has not been investigated. Using female mice deficient for SPOCK2, we assessed the role of SPOCK2 in influencing bone formation, the BMM and hematopoiesis. Using micro-computed tomography we found a significant decrease in trabecular bone volume, bone mineral density and thickness, but increased cortical mineral density in SPOCK2 knockout (KO) versus wildtype (WT) bones. C-terminal telopeptide of type I collagen, a measure of bone resorption, was significantly increased in bone marrow supernatants of SPOCK2 KO mice. In the hematopoietic compartment we found an increase in hematopoietic stem cells, but a decrease of mesenchymal stromal cells and adipocytes in the bone marrow of SPOCK2 KO mice compared to control mice. Megakaryocytes were increased in SPOCK2 KO mice. In summary, deficiency of SPOCK2 leads to several alterations in the BMM. The hematopoietic effects may be due to hematopoietic cell-intrinsic effects in SPOCK2-deficient cells or due to a SPOCK2-deficient niche or both.
Prognostic Value of CD8+ T Cells at the Invasive Margin Is Comparable to the Immune Score in Nonmetastatic Colorectal Cancer: A Prospective Multicentric Cohort Study
Wankhede D, Halama N , Kloor M, Edelmann D, Brenner H, Hoffmeister M
in: Clinical Cancer Research. 2025
Abstract
Purpose: The Immunoscore predicts colorectal cancer prognosis but faces adoption barriers because of complex software and reimbursement issues. This study used open-source methods to explore a simplified prognostic model in nonmetastatic colorectal cancer by focusing on single T-cell markers.
Experimental design: A multicentric prospective cohort study in patients with nonmetastatic colorectal cancer assessed CD3+ and CD8+ tumor-infiltrating lymphocytes (TIL) in the invasive margin (IM) and tumor core (TC) using QuPath. An immune cell score (ICS), based on TIL densities (CD3-IM, CD8-IM, CD3-TC, and CD8-TC), was calculated similarly to the Immunoscore. A split sample approach (70:30) estimated adjusted HRs for cancer-specific survival in training and validation sets. Classification and regression tree analysis identified the most prognostic TIL, and its model was compared with an ICS model for performance (Brier score) and discrimination (concordance probability estimate).
Results: Over a median follow-up of 9.0 years, 203 colorectal cancer-specific deaths occurred among 1,260 patients. Classification and regression tree-selected CD8-IM was the most prognostic TIL at a cutoff of 231 cells/mm2. Patients with high CD8-IM had better cancer-specific survival than low CD8-IM in both training (HR 0.58, 95% confidence interval, 0.40-0.84) and validation sets (HR 0.35, 95% confidence interval, 0.21-0.60). Brier scores of CD8-IM and ICS survival models were comparable in both training and validation cohorts, whereas the survival discrimination of CD8-IM slightly outperformed the ICS in the validation set (concordance probability estimate: CD8-IM: 0.748; ICS: 0.730).
Conclusions: CD8-IM alone provided prognostic information comparable with the ICS. Simplified, cost-effective TIL assessments could improve clinical translation and guide adjuvant therapy in early-stage colorectal cancer.
Rustims: An Open-Source Framework for Rapid Development and Processing of timsTOF Data-Dependent Acquisition Data
Teschner D, Gomez-Zepeda D, Łącki M K, Kemmer T, Busch A, Tenzer S, Hildebrandt A
in: Journal of Proteome Research. 2025
Abstract
Mass spectrometry is essential for analyzing and quantifying biological samples. The timsTOF platform is a prominent commercial tool for this purpose, particularly in bottom-up acquisition scenarios. The additional ion mobility dimension requires more complex data processing, yet most current software solutions for timsTOF raw data are proprietary or closed-source, limiting integration into custom workflows. We introduce rustims, a framework implementing a flexible toolbox designed for processing timsTOF raw data, currently focusing on data-dependent acquisition (DDA-PASEF). The framework employs a dual-language approach, combining efficient, multithreaded Rust code with an easy-to-use Python interface. This allows for implementations that are fast, intuitive, and easy to integrate. With imspy as its main Python scripting interface and sagepy for Sage search engine bindings, rustims enables fast, integrable, and intuitive processing. We demonstrate its capabilities with a pipeline for DDA-PASEF data including rescoring and integration of third-party tools like the Prosit intensity predictor and an extended ion mobility model. This pipeline supports tryptic proteomics and nontryptic immunopeptidomics data, with benchmark comparisons to FragPipe and PEAKS. Rustims is available on GitHub under the MIT license, with installation packages for multiple platforms on PyPi and all analysis scripts accessible via Zenodo.
Keywords: DDA-PASEF; Python; framework; ion mobility; mass spectrometry; open-source; proteomics; rust-lang; timsTOF.
The Chimeric Antigen Receptor T Cell Target Claudin 6 Is a Marker for Early Organ-Specific Epithelial Progenitors and Is Expressed in Some Pediatric Solid Tumor Entities
Seidmann L, Wingerter A, Metzig M O, Bornas A, El Malki K, Ustjanzew A, Ortmüller F, Kamyshanskiy Y, Kindler T, Laible M, Mohr X, Henninger N, Russo A, Beck O, Alt F, Wehling P, Roth W, Paret C, Faber J
in: Cancers. 2025
Abstract
Background/Objectives: The oncofetal membrane protein Claudin 6 (CLDN6) is an attractive target for T cell-based therapies. There is a lack of detailed analyses on the age-dependent expression of CLDN6 in normal tissues is lacking, which limits the expansion of CLDN6 CAR-T cell clinical trials to pediatric populations. Methods: We analyzed CLDN6 expression in extracranial solid tumors and normal tissues of children using RNA-sequencing data from over 500 pediatric solid tumor samples, qRT-PCR and immunohistochemistry (IHC) in more than 100 fresh-frozen tumor samples and, approximately, 250 formalin-fixed paraffin-embedded (FFPE) samples. We examined normal tissue expression via qRT-PCR in 32 different infant tissues and via IHC in roughly 290 tissues from donors across four age groups, as well as in fetal autopsy samples. Results: In fetal tissues, we detected CLDN6 expression primarily in the epithelial cells of several organs, including the skin, lungs, kidneys, intestinal tract, and pancreas, but not in undifferentiated blastemal cells. Postnatally, we found CLDN6-positive epithelial progenitors only during the first few weeks of life. In older-age groups, isolated clusters of CLDN6-positive progenitors were present, but in scarce quantities. In tumor tissues, we found strong and homogeneous CLDN6 expression in desmoplastic small round cell tumors and germ cell tumors. Wilms tumors demonstrated heterogeneous CLDN6 expression, notably absent in the blastemal component. Conclusions: These findings highlight an organ-specific presence of CLDN6-positive epithelial precursors that largely disappear in terminally differentiated epithelia within weeks after birth. Therefore, our data support CLDN6 as a viable therapeutic target in pediatric patients and justify their inclusion in basket studies for anti-CLDN6-based therapies.
Keywords: CAR-T cells; Claudin 6; Wilms tumors; desmoplastic small round cell tumors; germ cell tumors.
REDInet: a temporal convolutional network-based classifier for A-to-I RNA editing detection harnessing million known events
Fonzino A, Mazzacuva P L, Handen A, Silvestris D A, Arnold A, Pecori R, Pesole G, Picardi E
in: Briefings in Bioinformatics. 2025
Abstract
A-to-I ribonucleic acid (RNA) editing detection is still a challenging task. Current bioinformatics tools rely on empirical filters and whole genome sequencing or whole exome sequencing data to remove background noise, sequencing errors, and artifacts. Sometimes they make use of cumbersome and time-consuming computational procedures. Here, we present REDInet, a temporal convolutional network-based deep learning algorithm, to profile RNA editing in human RNA sequencing (RNAseq) data. It has been trained on REDIportal RNA editing sites, the largest collection of human A-to-I changes from >8000 RNAseq data of the genotype-tissue expression project. REDInet can classify editing events with high accuracy harnessing RNAseq nucleotide frequencies of 101-base windows without the need for coupled genomic data.
Keywords: A-to-I RNA editing; REDItools; RNAseq; temporal convolutional network.
Vaccine-induced T cell receptor T cell therapy targeting a glioblastoma stemness antigen
Chih Y-C, Dietsch A C, Koopmann P, Ma X, Agardy D A, Zhao B, Roia A, Kourtesakis A, Kilian M, Krämer C, Suwala A K, Stenzinger M, Boenig H, Blum A, Pienkowski V M,Aman K, Becker J P, Feldmann H, Bunse T, Harbottle R, Riemer A B, Liu H-K, Etminan N, Felix Sahm F, Bunse L.
in: Nature Communications. 2025
Abstract
T cell receptor-engineered T cells (TCR-T) could be advantageous in glioblastoma by allowing safe and ubiquitous targeting of the glioblastoma-derived peptidome. Protein tyrosine phosphatase receptor type Z1 (PTPRZ1), is a clinically targetable glioblastoma antigen associated with glioblastoma cell stemness. Here, we identify a therapeutic HLA-A*02-restricted PTPRZ1-reactive TCR retrieved from a vaccinated glioblastoma patient. Single-cell sequencing of primary brain tumors shows PTPRZ1 overexpression in malignant cells, especially in glioblastoma stem cells (GSCs) and astrocyte-like cells. The validated vaccine-induced TCR recognizes the endogenously processed antigen without off-target cross-reactivity. PTPRZ1-specific TCR-T (PTPRZ1-TCR-T) kill target cells antigen-specifically, and in murine experimental brain tumors, their combined intravenous and intracerebroventricular administration is efficacious. PTPRZ1-TCR-T maintain stem cell memory phenotype in vitro and in vivo and lyse all examined HLA-A*02+ primary glioblastoma cell lines with a preference for GSCs and astrocyte-like cells. In summary, we demonstrate the proof of principle to employ TCR-T to treat glioblastoma.
Neoadjuvant combination immunotherapy in MSI/dMMR colorectal cancer
Suarez-Carmona M, Halama N.
in: Trends in Cancer. 2024
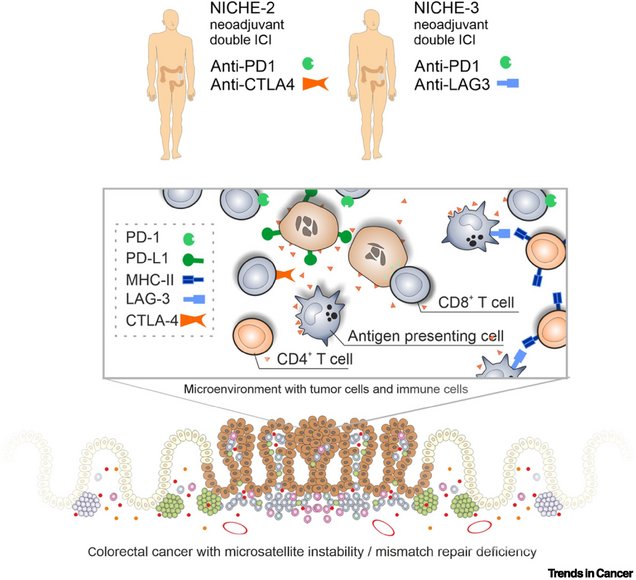 Trends in cancer
Trends in cancer
Abstract
Neoadjuvant immune checkpoint inhibition (ICI) is a new approach to treat patients with colorectal cancer (CRC). The effects of combined neoadjuvant ICI in locally advanced, DNA mismatch repair (dMMR)-deficient/microsatellite instable (MSI) CRC were recently reported by de Gooyer et al. from the NICHE-3 trial. Further studies will determine whether these impressive pathological responses lead to long-term clinical benefit.
Figure 1. Schematic overview of cells and immunomodulatory molecules within the tumor microenvironment of primary microsatellite instable (MSI) or DNA mismatch-repair (dMMR)-deficient colorectal cancer (with an enrichment of subsets of lymphocytes) and their specific inhibition in recent trials.
Abbreviations: ICI, immune checkpoint inhibition; MHC, major histocompatibility complex.
Keywords: MSI; colorectal cancer; dMMR; immune checkpoint inhibition; neoadjuvant therapy.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
optiPRM: A targeted immunopeptidomics LC-MS workflow with ultra-high sensitivity for the detection of mutation-derived tumor neoepitopes from limited input material
Salek M, Förster J D, Becker J P, Meyer M, Charoentong P, Lyu Y, Lindner K, Lotsch C, Volkmar M, Momburg F, Poschke I, Fröhling S, Schmitz M, Offringa R, Platten M, Jäger D, Zörnig I, Riemer A B.
in: Molecular & Cellular Proteomics. 2024
Abstract
Personalized cancer immunotherapies such as therapeutic vaccines and adoptive transfer of T cell receptor (TCR)-transgenic T cells rely on the presentation of tumor-specific peptides by human leukocyte antigen (HLA) class I molecules to cytotoxic T cells. Such neoepitopes can for example arise from somatic mutations and their identification is crucial for the rational design of new therapeutic interventions. Liquid chromatography mass spectrometry (LC-MS)-based immunopeptidomics is the only method to directly prove actual peptide presentation and we have developed a parameter optimization workflow to tune targeted assays for maximum detection sensitivity on a per peptide basis, termed optiPRM. Optimization of collision energy using optiPRM allows for improved detection of low abundant peptides that are very hard to detect using standard parameters. Applying this to immunopeptidomics, we detected a neoepitope in a patient-derived xenograft (PDX) from as little as 2.5×106 cells input. Application of the workflow on small patient tumor samples allowed for the detection of five mutation-derived neoepitopes in three patients. One neoepitope was confirmed to be recognized by patient T cells. In conclusion, optiPRM, a targeted MS workflow reaching ultra-high sensitivity by per peptide parameter optimization, which makes the identification of actionable neoepitopes possible from sample sizes usually available in the clinic.
© 2024. The Author(s).
Sphinganine recruits TLR4 adaptors in macrophages and promotes inflammation in murine models of sepsis and melanoma
Hering M, Madi A, Sandhoff R, Ma S, Wu J, Mieg A, Richter K, Mohr K, Knabe N. Stichling D, Poschet G, Bestvater F, Frank L, Utikal J, Umansky V, Cui G.
in: Nature Communications. 2024
 BioRender.com
BioRender.com
Abstract
After recognizing its ligand lipopolysaccharide, Toll-like receptor 4 (TLR4) recruits adaptor proteins to the cell membrane, thereby initiating downstream signaling and triggering inflammation. Whether this recruitment of adaptor proteins is dependent solely on protein-protein interactions is unknown. Here, we report that the sphingolipid sphinganine physically interacts with the adaptor proteins MyD88 and TIRAP and promotes MyD88 recruitment in macrophages. Myeloid cell-specific deficiency in serine palmitoyltransferase long chain base subunit 2, which encodes the key enzyme catalyzing sphingolipid biosynthesis, decreases the membrane recruitment of MyD88 and inhibits inflammatory responses in in vitro bone marrow-derived macrophage and in vivo sepsis models. In a melanoma mouse model, serine palmitoyltransferase long chain base subunit 2 deficiency decreases anti-tumor myeloid cell responses and increases tumor growth. Therefore, sphinganine biosynthesis is required for the initiation of TLR4 signal transduction and serves as a checkpoint for macrophage pattern recognition in sepsis and melanoma mouse models.
© 2024. The Author(s).
The immunosuppressive drug cyclosporin A has an immunostimulatory function in CD8+ T cells
Wißfeld J, Hering M, ten Bosch N, Cui G.
in: European Journal of Immunology
Abstract
Cyclosporin A is a well-established immunosuppressive drug used to treat or prevent graft-versus-host disease, the rejection of organ transplants, autoimmune disorders, and leukemia. It exerts its immunosuppressive effects by inhibiting calcineurin-mediated dephosphorylation of the nuclear factor of activated T cells (NFAT), thus preventing its nuclear entry and suppressing T cell activation. Here we report an unexpected immunostimulatory effect of cyclosporin A in activating the mammalian target of rapamycin complex 1 (mTORC1), a crucial metabolic hub required for T cell activation. Through screening a panel of tool compounds known to regulate mTORC1 activation, we found that cyclosporin A activated mTORC1 in CD8+ T cells in a 3-phosphoinositide-dependent protein kinase 1 (PDK1) and protein kinase B (PKB/AKT)-dependent manner. Mechanistically, cyclosporin A inhibited the calcineurin-mediated AKT dephosphorylation, thereby stabilizing mTORC1 signaling. Cyclosporin A synergized with mTORC1 pathway inhibitors, leading to potent suppression of proliferation and cytokine production in CD8+ T cells and an increase in the killing of acute T cell leukemia cells. Consequently, relying solely on CsA is insufficient to achieve optimal therapeutic outcomes. It is necessary to simultaneously target both the calcineurin-NFAT pathway and the mTORC1 pathway to maximize therapeutic efficacy.
T-Cell-Based Platform for Functional Screening of T-Cell Receptors Identified in Single-Cell RNA Sequencing Data Sets of Tumor-Infiltrating T-Cells
Rodriguez Ehrenfried A, Zens S, Steffens LK, Kehm H, Paul A, Lauenstein C, Volkmar M, Poschke I, Meng Z, Offringa R.
in: Bio-protocol. 2024
Abstract
The advent of single-cell RNA sequencing (scRNAseq) has enabled in-depth gene expression analysis of several thousand cells isolated from tissues. We recently reported the application of scRNAseq toward the dissection of the tumor-infiltrating T-cell repertoire in human pancreatic cancer samples. In this study, we demonstrated that combined whole transcriptome and T-cell receptor (TCR) sequencing provides an effective way to identify tumor-reactive TCR clonotypes on the basis of gene expression signatures. An important aspect in this respect was the experimental validation of TCR-mediated anti-tumor reactivity by means of an in vitro functional assay, which is the subject of the present protocol. This assay involves the transient transfection of mRNA gene constructs encoding TCRα/β pairs into a well-defined human T-cell line, followed by co-cultivation with the tumor cells of interest and detection of T-cell activation by flow cytometry. Due to the high transfectability and the low background reactivity of the mock-transfected T-cell line to a wide variety of tumor cells, this assay offers a highly robust and versatile platform for the functional screening of large numbers of TCR clonotypes as identified in scRNAseq data sets. Whereas the assay was initially developed to test TCRs of human origin, it was more recently also applied successfully for the screening of TCRs of murine origin. Key features • Efficient functional screening of-and discrimination between-TCRs isolated from tumor-reactive vs. bystander T-cell clones. • Applicable to TCRs from CD8+ and CD4+ tumor-infiltrating T-cells originating from patient-derived tumor samples and syngeneic mouse tumor models. • Rapid flow cytometric detection of T-cell activation by means of TNFα and CD107a expression after a 5 h T-cell/tumor cell co-cultivation.
Prediction of tumor-reactive T cell receptors from scRNA-seq data for personalized T cell therapy
Tan CL, Lindner K, Boschert T, Meng Z, Rodriguez Ehrenfried A, De Roia A, Haltenhof G, Faenza A, Imperatore F, Bunse L, Lindner JM, Harbottle RP, Ratliff M, Offringa R, Poschke I, Platten M, Green EW
in: Nature Biology. 2024
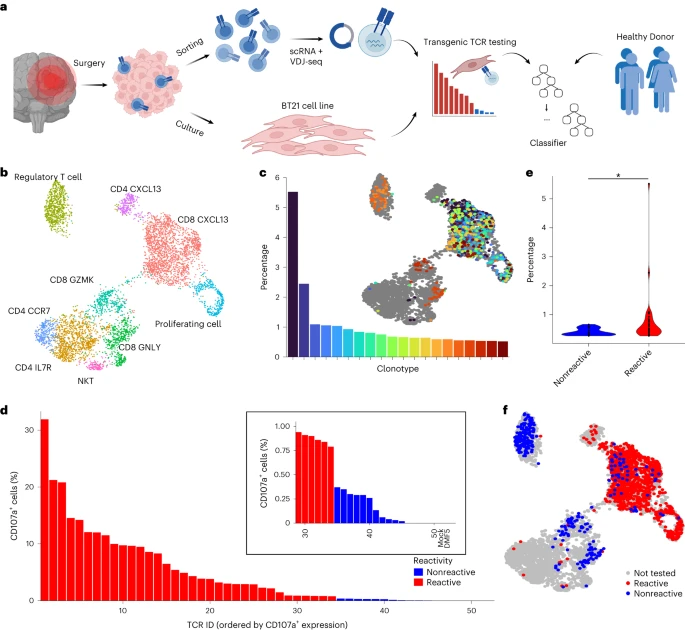
Abstract
The identification of patient-derived, tumor-reactive T cell receptors (TCRs) as a basis for personalized transgenic T cell therapies remains a time- and cost-intensive endeavor. Current approaches to identify tumor-reactive TCRs analyze tumor mutations to predict T cell activating (neo)antigens and use these to either enrich tumor infiltrating lymphocyte (TIL) cultures or validate individual TCRs for transgenic autologous therapies. Here we combined high-throughput TCR cloning and reactivity validation to train predicTCR, a machine learning classifier that identifies individual tumor-reactive TILs in an antigen-agnostic manner based on single-TIL RNA sequencing. PredicTCR identifies tumor-reactive TCRs in TILs from diverse cancers better than previous gene set enrichment-based approaches, increasing specificity and sensitivity (geometric mean) from 0.38 to 0.74. By predicting tumor-reactive TCRs in a matter of days, TCR clonotypes can be prioritized to accelerate the manufacture of personalized T cell therapies.
a: An overview of the experimental and computational pipeline underlying the predicTCR classifier: TILs are sorted and subject to scRNA + VDJ-seq, while adjacent resected tumor material is used to establish the BT21 tumor cell line. TCR reactivity data are then integrated with scRNA + VDJ-seq data to train the predicTCR classifier, which is later tested on externally generated TIL datasets from diverse tumor types.
b: Unsupervised clustering (UMAP plot) of scRNA-seq data of TILs (n = 5,651) recovered from brain metastasis sample, with key T cell subtypes annotated.
c: The percentage frequency of the top 20 TIL TCR clonotypes and their distribution projected onto the UMAP, showing that cells of the same clonotype can occupy diverse phenotypic states.
d: T cells transfected with one of the 50 most frequently occurring TIL-derived TCR clonotypes (representing 58 distinct TCR α/ß chain pairs) are cocultured with BT21 cells; the resulting levels of CD107a (as quantified by flow cytometry, gated on mTCRβ+ cells, which express the transgenic TCR as a chimera with the murine constant domain) demonstrate whether a given TCR clonotype recognizes the BT21 cell line. For details of settings per TCR reactivity threshold, see Methods. DMF5 is the HLA mismatched negative control TCR.
e: BT21-reactive TCR clonotypes are more frequent than nonreactive clonotypes in the TIL population.
f: BT21 reactivity testing results projected onto the UMAP plot (b).
T-FINDER: A highly sensitive, pan-HLA platform for functional T cell receptor and ligand discovery
Cetin M, Pinamonti V, Schmid T, Boschert T, Mellado Fuentes A, Kromer K, Lerner T, Zhang J, Herzig Y, Ehlert C, Hernandez-Hernandez M, Samaras G, Maldonado Torres C, Fisch L, Dragan V, Kouwenhoven A, Van Schoubroeck B, Wils H, Van Hove C, Platten M, Green EW, Stevenaert F, Felix NJ, Lindner JM.
in: Science Advances. 2024
Abstract
Effective, unbiased, high-throughput methods to functionally identify both class II and class I HLA-presented T cell epitopes and their cognate T cell receptors (TCRs) are essential for and prerequisite to diagnostic and therapeutic applications, yet remain underdeveloped. Here, we present T-FINDER [T cell Functional Identification and (Neo)-antigen Discovery of Epitopes and Receptors], a system to rapidly deconvolute CD4 and CD8 TCRs and targets physiologically processed and presented by an individual's unmanipulated, complete human leukocyte antigen (HLA) haplotype. Combining a highly sensitive TCR signaling reporter with an antigen processing system to overcome previously undescribed limitations to target expression, T-FINDER both robustly identifies unknown peptide:HLA ligands from antigen libraries and rapidly screens and functionally validates the specificity of large TCR libraries against known or predicted targets. To demonstrate its capabilities, we apply the platform to multiple TCR-based applications, including diffuse midline glioma, celiac disease, and rheumatoid arthritis, providing unique biological insights and showcasing T-FINDER's potency and versatility.
H3K27M neoepitope vaccination in diffuse midline glioma induces B and T cell responses across diverse HLA loci of a recovered patient
Boschert T, Kromer K, Lerner T, Lindner K, Haltenhof G, Tan CL, Jähne K, Poschke I, Bunse L, Eisele P, Grassl N, Mildenberger I, Sahm K, Platten M, Lindner JM, Green EW.
In: Science Advances. 2024
Abstract
H3K27M, a driver mutation with T and B cell neoepitope characteristics, defines an aggressive subtype of diffuse glioma with poor survival. We functionally dissect the immune response of one patient treated with an H3K27M peptide vaccine who subsequently entered complete remission. The vaccine robustly expanded class II human leukocyte antigen (HLA)-restricted peripheral H3K27M-specific T cells. Using functional assays, we characterized 34 clonally unique H3K27M-reactive T cell receptors and identified critical, conserved motifs in their complementarity-determining region 3 regions. Using detailed HLA mapping, we further demonstrate that diverse HLA-DQ and HLA-DR alleles present immunogenic H3K27M epitopes. Furthermore, we identified and profiled H3K27M-reactive B cell receptors from activated B cells in the cerebrospinal fluid. Our results uncover the breadth of the adaptive immune response against a shared clonal neoantigen across multiple HLA allelotypes and support the use of class II-restricted peptide vaccines to stimulate tumor-specific T and B cells harboring receptors with therapeutic potential.
Thunder-DDA-PASEF enables high-coverage immunopeptidomics and is boosted by MS2Rescore with MS2PIP timsTOF fragmentation prediction model
Gomez-Zepeda D, Arnold-Schild D, Beyrle J, Declercq A, Gabriels R, Kumm E, Preikschat A, Łącki MK, Hirschler A, Rijal JB, Carapito C, Martens L, Distler U, Schild H, Tenzer S.
In: Nature Communications. 2024
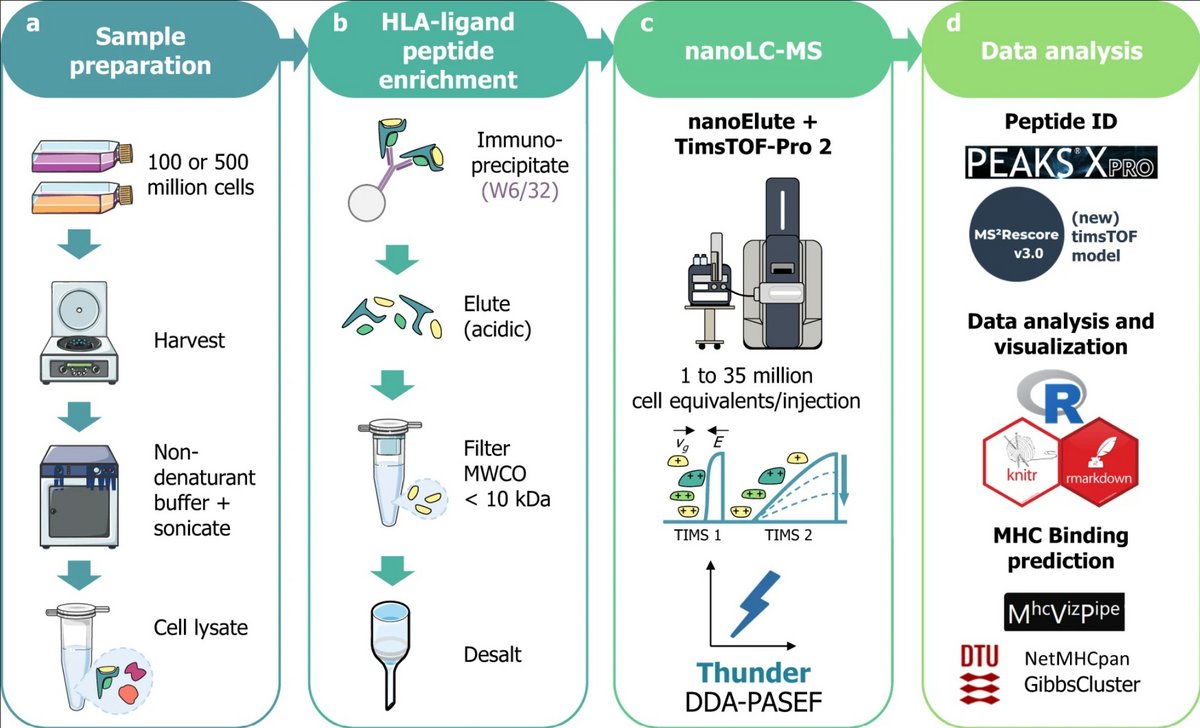
Abstract
Human leukocyte antigen (HLA) class I peptide ligands (HLAIps) are key targets for developing vaccines and immunotherapies against infectious pathogens or cancer cells. Identifying HLAIps is challenging due to their high diversity, low abundance, and patient-specific profiles. Here, we developed a highly sensitive method for identifying HLAIps using liquid chromatography-ion mobility-tandem mass spectrometry (LC-IMS-MS/MS). The optimized method, Thunder-DDA-PASEF, semi-selectively fragments HLAIps based on their IMS and m/z, thus increasing the coverage of immunopeptidomics analyses. Thunder-DDA-PASEF includes singly-charged peptides, which contributes to more than 35% of the HLAIp identifications. Combined with MS2Rescore, Thunder-DDA-PASEF improved ligandome coverage by 150% compared to the original-DDA-PASEF method, and enabled in-depth profiling of HLAIps from two human cell lines, JY and Raji, transfected to express the SARS-CoV-2 spike protein. We identified seventeen spike protein HLAIps, thirteen of which had been reported to elicit immune responses in human patients.
a: Sample preparation: 100 or 500 million cells of diverse cell lines were harvested, then lysed by sonication in 1% CHAPS in PBS buffer (m/v). Alternatively, 4 mL of unfractionated plasma were processed.
b: HLA-ligand peptide enrichment: was performed by immunoaffinity using the W6/32 anti-HLA-A, B, C antibody coupled to CNBr-activated agarose beads; after overnight incubation and several washes, peptides were eluted with 0.2% trifluoro-acetic acid (v/v), ultrafiltered on molecular weight cutoff filters (MWCO, 10 kDa cutoff) and desalted in HLB plates (Waters Corp.).
c: NanoLC-MS: analysis was performed using a nanoElute coupled to timsTOF-Pro-2 in DDA-PASEF18 with different parameters to optimize the MS acquisition.
d: Data analysis: Database search was performed in PEAKS XPro using unspecific cleavage. After training a MS2PIP (Declercq, A. et al. Updated MS2PIP web server supports cutting-edge proteomics applications. Nucleic Acids Res. 51, W338–W342 (2023).)timsTOF fragmentation prediction model, peptide identification was rescored using MS2Rescore (MS2R, v3.0.0b4) (Declercq, A. et al. MS2Rescore: data-driven rescoring dramatically boosts immunopeptide identification rates. Mol. Cell. Proteom. 21, 100266 (2022).; Buur, L. M. et al. MS2Rescore 3.0 is a modular, flexible, and user-friendly platform to boost peptide identifications, as showcased with MS Amanda 3.0. Preprint available at ChemRxiv 1–23 (2023).). Data analysis was performed in R and predicted MHC-binding affinity was evaluated using NetMHCpan-4.1 (Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2021).) and GibbsCluster-2.0 (Andreatta, M., Alvarez, B. & Nielsen, M. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res. 45, W458–W463 (2017).) through MhcVizPipe (v0.7.9) (Kovalchik, K. A. et al. MhcVizPipe: a quality control software for rapid assessment of small- to large-scale immunopeptidome datasets. Mol. Cell. Proteom. 21, 0–14 (2022). ).
Metabolic regulation of immune responses to cancer
Wißfeld J*, Werner A*, Yan X, Ten Bosch N, Cui G. *Authors contributed equally.
In: Cacer Biology & Medicine
The tumor microenvironment is an ecosystem composed of multiple types of cells, such as tumor cells, immune cells, and cancer-associated fibroblasts. Cancer cells grow faster than non-cancerous cells and consume larger amounts of nutrients. The rapid growth characteristic of cancer cells fundamentally alters nutrient availability in the tumor microenvironment and results in reprogramming of immune cell metabolic pathways. Accumulating evidence suggests that cellular metabolism of nutrients, such as lipids and amino acids, beyond being essential to meet the bioenergetic and biosynthetic demands of immune cells, also regulates a broad spectrum of cellular signal transduction, and influences immune cell survival, differentiation, and anti-tumor effector function. The cancer immunometabolism research field is rapidly evolving, and exciting new discoveries are reported in high-profile journals nearly weekly. Therefore, all new findings in this field cannot be summarized within this short review. Instead, this review is intended to provide a brief introduction to this rapidly developing research field, with a focus on the metabolism of two classes of important nutrients-lipids and amino acids-in immune cells. We highlight recent research on the roles of lipids and amino acids in regulating the metabolic fitness and immunological functions of T cells, macrophages, and natural killer cells in the tumor microenvironment. Furthermore, we discuss the possibility of "editing" metabolic pathways in immune cells to act synergistically with currently available immunotherapies in enhancing anti-tumor immune responses.
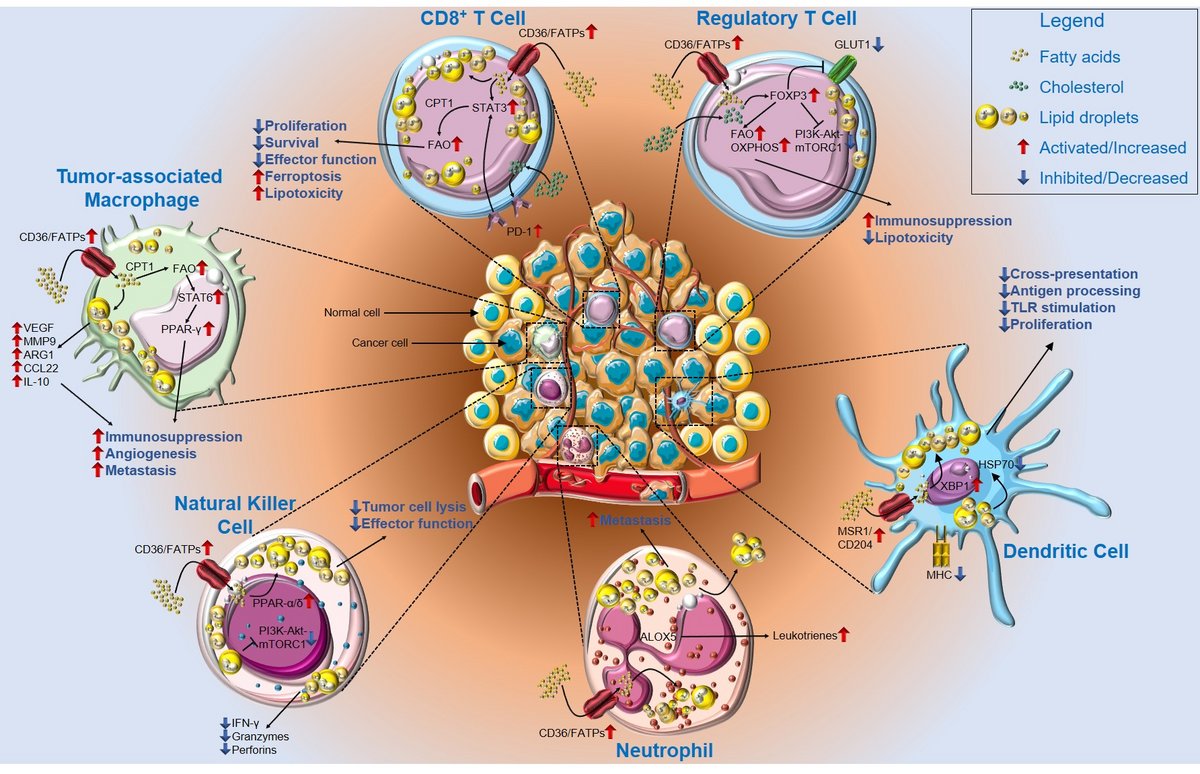
Figure 1: Lipid metabolism impairs anti-tumor immunity. In the lipid-rich TME, infiltrating CD8+ T cells upregulate CD36 or FATPs, thereby increasing lipid uptake. Lipid accumulation and subsequent storage in lipid droplets results in a metabolic switch toward fatty acid oxidation (FAO) via STAT3 and CPT1, thus decreasing CD8+ T cell proliferation, survival, and overall effector function, but increasing susceptibility to ferroptosis and lipotoxicity. In contrast, Treg cells use FAO and oxidative phosphorylation to sustain their immunosuppressive phenotype in a FoxP3-dependent manner through downregulation of GLUT1 and the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin complex 1 (mTORC1) pathway.
Lipid accumulation in DCs is mediated by MRS1/CD204 as well as in an XBP1-dependent manner, and it interferes with TLR stimulation and proliferation of DCs. Furthermore, accumulated lipids impair antigen processing and cross-presentation by HSP70 resorption. Neutrophils accumulate lipids via CD36/FATPs and produce leukotrienes via ALOX5. Lipid droplets are then transferred to metastasis-initiating tumor cells, where they facilitate survival. Natural killer (NK) cells show an impaired metabolic profile characterized by PPAR-α/δ-driven lipid accumulation and a decrease in phosphoinositide 3-kinase-Akt-mTORC1 signaling. This metabolic shift results in decreased secretion of effector cytokines, granzymes, and perforins, as well as decreased tumor cell lysis. Tumor-associated macrophages (TAMs) increase lipid uptake and storage, as well as PPAR-γ signaling via FAO and STAT6, thus increasing the secretion of tumor promoting and anti-inflammatory factors, and supporting angiogenesis and metastasis. Abbreviations: Akt, Akt serine/threonine kinase 1; ALOX5, arachidonate 5-lipoxygenase; ARG1, arginase 1; CCL22, chemokine (C-C motif) ligand 22; CD, cluster of differentiation; CPT1, carnitine palmitoyltransferase 1; DC, dendritic cell; FAO, fatty acid oxidation; FATP, fatty acid transport protein; FoxP3, forkhead box P3; GLUT1, glucose transporter 1; HSP70, heat shock protein 70 kDa; IFN-γ, interferon γ; IL, interleukin; MHC, major histocompatibility complex; MMP9, matrix metallopeptidase 9; MRS1, macrophage scavenger receptor 1; mTORC1, mechanistic target of rapamycin kinase complex 1; NO, nitric oxide; PD-1, programmed cell death protein 1; PD-L1, programmed cell death 1 ligand 1; PPAR, peroxisome proliferator activated receptor; STAT, signal transducer and activator of transcription; TAM, tumor-associated macrophage; TME, tumor microenvironment; Treg cells, regulatory T cells; XBP1, X-box binding protein 1.
Parts of the figure were drawn by using original or modified pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License